We aim to give people back their lives,
using Hyperbaric Oxygen Therapy.
treating over 100 different conditions
The mission of Harch Hyperbarics, Inc. is to maximize health and well-being through the personalized application of hyperbaric oxygen therapy. Harch Hyperbarics intends to shift the paradigm to treatment of neurological, acute, \ and chronic diseases from one of diagnosis and observation. Since 1986 we have used HBOT’s gene and stem cell stimulating properties to “give people back their lives.” Harch Hyperbarics’ goal is to expand this therapy to elevate global health and longevity.
WHAT YOU NEED TO KNOW:
Individual dosing of HBOT for neurological conditions with quantitative electroencephalogram (qEEG)
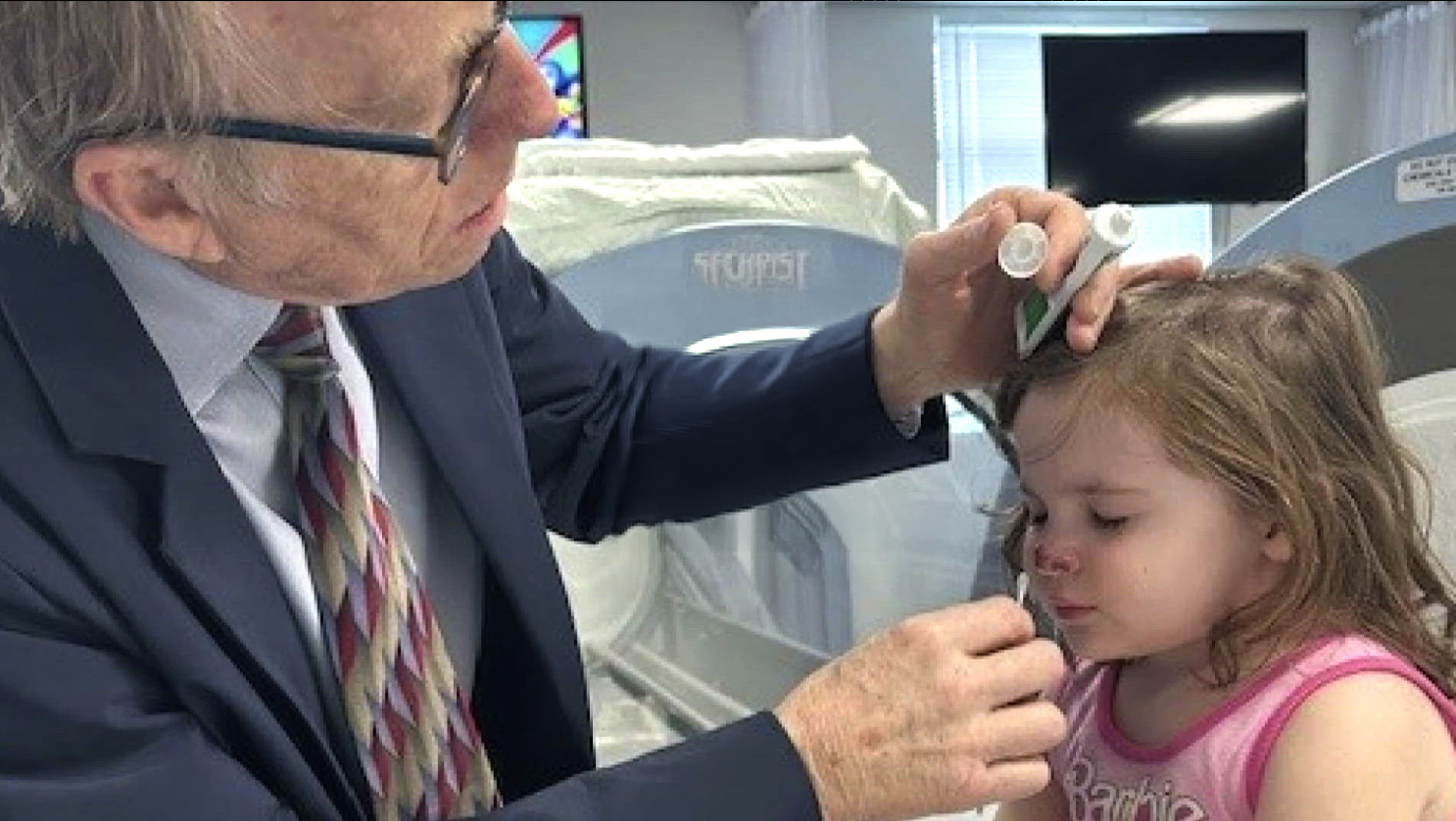
Harch Hyperbarics offers precision-dosing of hyperbaric oxygen therapy customized to the patient, the patient’s condition, and the time of intervention in the disease process (days to years after the onset of the disease). This is done in hardshell chambers with the entire dosing range of pressures, and oxygen content.
For patients evaluated and treated in New Orleans who have demonstrated responsiveness to HBOT and require additional HBOT, but cannot return to New Orleans for further treatment Harch Hyperbarics offers Long-Term HBOT in your home. We will install a chamber and train family members to treat the patient in a home setting.
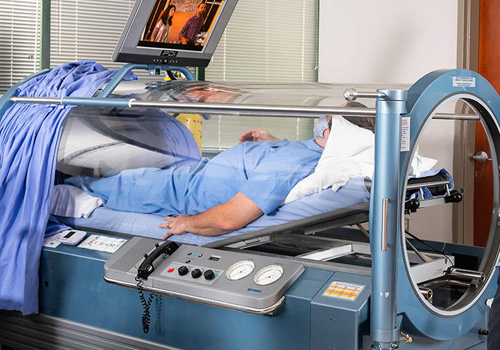
Exclusive service is for patients who want the convenience of personalized in-home treatment by a trained technician or those who cannot travel for treatment to New Orleans and require or request technician-delivered HBOT. It is a turnkey service from consultation, evaluation, personalized dosing and more.
About Dr. Paul Harch
Leading hyperbaric medicine, diving, and emergency medicine physician
Patients travel from countries all over the world to receive hyperbaric oxygen treatment with Dr. Harch. Dr. Harch has successfully treated patients with a wide range of diagnoses from drowned children to U.S. servicemen with traumatic brain injury (TBI) and post-traumatic stress disorder (PTSD). He has presented his experience to the U.S. Congress four times and is the co-author of the book “The Oxygen Revolution.”
LEARNING ABOUT
Hyperbaric oxygen therapy
THE BEST WAY TO HELP YOU IS TO INFORM YOU
Hyperbaric oxygen therapy, or HBOT, is a medical treatment that enhances the body’s natural healing processes. It is a simple, non-invasive, and painless treatment. The combination of increased atmospheric pressure and increased percentage of oxygen in a total body chamber treats a wide variety of medical conditions. It is also commonly referred to as oxygen treatment, hyperbaric therapy, or hyperbaric oxygen treatment.
Under normal circumstances, oxygen is transported throughout the body by red blood cells with a tiny amount also dissolved in plasma, the liquid portion of the blood. When oxygen is delivered under pressure much greater amounts of oxygen are dissolved in plasma where it is more readily available for transfer to all of the tissues and fluids in the body, such as the central nervous system fluid, the lymphatic system, connective tissue, all solid organs, and bone. In the smallest blood vessels, plasma blood flow carries the increased oxygen to areas where circulation is diminished or blocked. In this way, adequate oxygen can reach all of the damaged tissues, especially those where healing is delayed or diminished. Once sufficient oxygen is delivered to these damaged tissues, the oxygen reduces swelling. This greatly enhances the ability of white blood cells to kill bacteria and stimulates new blood vessels to grow more rapidly into the affected areas.


There are many benefits of HBOT. Healing cannot take place without appropriate oxygen levels in the tissue and cells. Many illnesses and injuries fail to heal because of inadequate oxygen levels. Increased oxygen under pressure, or HBOT, provides this extra oxygen to tissue and cells with minimal side effects. Some of the many benefits of hyperbaric oxygen therapy include:
- New connective tissue growth
- New blood vessel growth
- New skin growth
- Remodeling of bone
- Expedited healing
- New cell regeneration
- Anti-aging
- A feeling of well being
- Improved quality of life
Potentially, HBOT can be used for any condition that involves injury, wounding, loss of blood supply, or inflammation. At Harch HBOT, we treat more than 70 conditions with hyperbaric oxygen therapy. The following are some of the most common conditions and injuries that Dr. Harch treats with oxygen therapy.
- Autism
- Cerebral Palsy
- Drowning and Anoxic Brain Injury
- Migraine Headache
- Mild Cognitive Impairment (MCI) and Dementia
- Multiple Sclerosis
- Plastic-Cosmetic Surgery
- Recovery from Surgery
- Sports Injuries and Sports Performance/Recovery
- Stroke
- Traumatic Brain Injury
- And many more
Even if the condition or injury that you or a loved one is suffering from is not listed here, it does not mean that it is not responsive to HBOT or that we cannot successfully treat you with hyperbaric oxygen therapy. Hyperbaric oxygen therapy is different from all other known therapies in that it treats the underlying disease processes that are responsible for and common to many diseases. Diseases that are similar to those diseases that are typically treated with HBOT are also very often responsive to HBOT. Please contact us today to find out if you or your loved one is a good candidate for HBOT with Dr. Harch.
SEARCH FOR YOUR CONDITION:
HBOT is most recently being used to treat COVID-19 and appears to be working well for most coronavirus cases. In early 2020, Dr. Harch announced to the medical community in a post that “History bears remembering.” He noted that the introduction of HBOT in the United States by Dr. Orval Cunninghan in 1918 was to Spanish Flu patients dying of the same lung condition as COVID-19 patients. At the same time, Dr. Xaioling Zhong in Wuhan successfully treated 35 patients with COVID-19, six of whom were deteriorating. Since then, multiple centers have reported the beneficial impact of HBOT on dying COVID-19 patients. Dr. Harch has been successfully treating patients with COVID-19 Long-Haulers Syndrome. Contact us for more details about how HBOT can benefit patients affected by COVID-19.
A hyperbaric chamber is a fully enclosed vessel in which increased pressure and increased amounts of oxygen are therapeutically delivered. At Harch HBOT, we use state-of-the-art, monoplace chambers of clear acrylic which have been customized to administer the entire range of clinical hyperbaric oxygen therapy doses. During a treatment, the increased air pressure in the chamber causes greater than normal amounts of oxygen to dissolve in the blood passing through the lungs. The increased pressure itself also has beneficial effects for patients. Our clear chambers allow our professionally trained technicians to closely monitor the patient and allow the patient to see outside the chamber easily. Patients are able to communicate with the attending technician via an intercom, and they can watch a movie, listen to music, or rest during their treatment.



Overall, hyperbaric oxygen therapy is extremely safe. Hyperbaric oxygen therapy is prescribed by a physician and performed under medical supervision. It is non-invasive, painless, and helps with a wide variety of medical conditions and injuries. However, like all medical treatments, there are some minor risks. The risks will be discussed with you before you sign your consent form for therapy.
HBOT Success Stories
Here are the stories of our incredible patients.

This is the story of Eden Carlson, a 2-year old girl who experienced cardiac arrest after cold water drowning. She technically died for nearly drowning. After hyperbaric treatments with Dr. Harch, Eden regained normal speech and cognition, assisted gait, residual fine motor and temperament deficits.

Curt Allen, Jr. was a 17 year old man involved in a high speed motor vehicle accident in which he sustained severe traumatic brain injury. After hyperbaric treatments he is now employed, walking and talking again. Learn More

3 year-old Robert Boytim was found unresponsive in a pond. After an hour of CPR, EMS got a heartbeat. The road to recovery began. When Robert visited Dr. Harch his spine was curved completely backwards. Two days after his first treatment, his bend was gone.